Cellular Immunology Section
Ethan M. Shevach, M.D.
Chief, Cellular Immunology Section

Major Areas of Research
- We are studying the role of NK, Antigen Presenting Cells (APC) and Tregs in the immune system homeostasis in the steady-state and disease, especially the role of Ly49s or LILRBs/MHC-I and TCR/MHC-II interactions in viral and tumor immunity.
- We are investigating mechanisms of Treg mediated suppression. Especially a Treg mediated process similar to trogocytosis to remove peptide MHC-II complexes from dendritic cells (DC) rendering the DC incompetent to present the targeted pMHC-II complex.
- We are interested in elucidating the function of two members of the Ikaros gene transcription factor (TF) family, Helios and Eos. Both of these TFs are preferentially expressed in Treg and play roles in controlling certain aspects of Treg suppressor functions.
- We are studying the role of type I interferons (IFNs) in Treg function. We had previously demonstrated that type I IFNs play a role in Treg development and survival.
- We are working on bringing some of our discoveries from the bench to the clinic by studying hTregs biology by using immunodeficient mice engrafted with human peripheral blood mononuclear cells in collaborations with industry.
Program Description
The major focus of the Cellular Immunology Section over the past 25 years has been furthering our understanding of the function of T regulatory cells (Tregs) that express the transcription factor Foxp3. Our group was one of the first in the world to realize the importance of Treg and we performed many of the initial studies that described their phenotype and function. The study of Tregs is now one of the most active areas of research in basic and clinical immunology and the therapeutic use of Treg is now in the clinic. It was originally assumed that Treg were a dedicated lineage of cells that developed only from thymic precursors, but more recent studies have clearly documented that Foxp3+ T cells also develop from conventional T cells (Tconv) in extra-thymic peripheral sites in vivo, and these are termed peripherally induced Tregs (pTregs). Treg can also be generated in culture in the presence of transforming growth factor-b1 (TGF-b1) and are termed induced Tregs (iTreg). The relative importance of tTreg and pTreg is unknown.
Although most of our studies deal with Tregs in mouse models, over the past 15 years we have also carried out studies on human Tregs (hTregs) derived from normal donors. Our ongoing studies are described below and have been divided up into five major projects with significant overlap between the projects. We also intentionally validate new and novel findings that we learn in one species with similar studies in another species, as there appears to be conservation of many aspects of Treg function across the species.
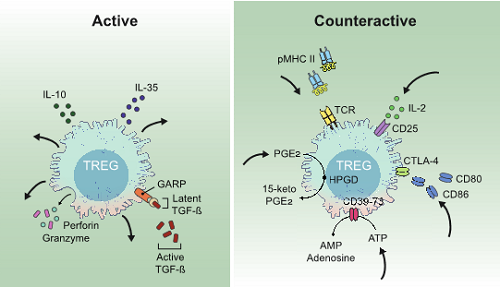
Two fundamentally different suppressor modes characterize Treg suppressor mechanisms. In the “active” mode, Treg cells secrete immunoinhibitory molecules that exert their effects on other cell types. In contrast, in the “counteractive” mode Treg cells are actively engaged in removing vital components from other cells types including antigen, costimulatory molecules, cytokines, and inflammatory signals thereby decreasing the activation of T effector cells. B. Akkaya and E.M. Shevach, Cellular immunology 2020.
Biography
Education
M.D., 1967, Boston University
Dr. Shevach received his M.D. from Boston University in 1967. Following clinical training, he joined the Laboratory of Immunology as a senior staff fellow in 1972, was appointed a senior investigator in 1973, and became a section chief in 1987. Dr. Shevach served as editor-in-chief of the Journal of Immunology from 1987 to 1992 and editor-in-chief of Cellular Immunology from 1996 to 2007. Dr. Shevach is the author of more than 450 papers.
Awards
Distinguished Service Award (The American Association of Immunologists)
Distinguished Alumnus Award (Boston University School of Medicine)
2004 William B. Coley Award for Distinguished Research in Basic and Tumor Immunology
Distinguished Scientist Award, World Allergy Congress, 2015
Thompson-Reuters Citation Laureate, 2015
Distinguished Fellow, American Association of Immunologists, 2019
Memberships
The American Association of Immunologists
American Society for Clinical Investigation
Association of American Physicians
Editorial Boards
Immunity
The Journal of Immunological Methods
Journal of Biomedical Science
Journal of Experimental Medicine
Current Protocols in Immunology
Human Immunology
Selected Publications
Tanwar S, Oguz C, Metidji A, Dahlstrom E, Barbian K, Kanakabandi K, Sykora L, Shevach EM. Type I IFN signaling in T regulatory cells modulates chemokine production and myeloid derived suppressor cells trafficking during EAE. J Autoimmun. 2020 Jul 21;102525. Epub ahead of print.
Panda AK, Gangaplara A, Buszko M, Natarajan K, Boyd LF, Sharma S, Margulies DH, Shevach EM. Cutting Edge: Inhibition of the Interaction of NK Inhibitory Receptors with MHC Class I Augments Antiviral and Antitumor Immunity. J Immunol. 2020 Aug 1;205(3):567-572.
Lopez-Ocasio M, Buszko M, Blain M, Wang K, Shevach EM. T Follicular Regulatory Cell Suppression of T Follicular Helper Cell Function Is Context-Dependent in vitro. Front Immunol. 2020 Apr 17;11:637.
Gokhale AS, Gangaplara A, Lopez-Occasio M, Thornton AM, Shevach EM. Selective deletion of Eos (Ikzf4) in T-regulatory cells leads to loss of suppressive function and development of systemic autoimmunity. J Autoimmun. 2019 Dec;105:102300.
Skadow M, Penna VR, Galant-Swafford J, Shevach EM, Thornton AM. Helios Deficiency Predisposes the Differentiation of CD4+Foxp3- T Cells into Peripherally Derived Regulatory T Cells. J Immunol. 2019 Jul 15;203(2):370-378.
Akkaya B, Oya Y, Akkaya M, Al Souz J, Holstein AH, Kamenyeva O, Kabat J, Matsumura R, Dorward DW, Glass DD, Shevach EM. Regulatory T cells mediate specific suppression by depleting peptide-MHC class II from dendritic cells. Nat Immunol. 2019 Feb;20(2):218-231.
Research Group
The major focus of the Cellular Immunology Section over the past 25 years has been furthering our understanding of the function of T regulatory cells (Tregs) that express the transcription factor Foxp3. Our group was one of the first in the world to realize the importance of Treg and we performed many of the initial studies that described their phenotype and function.
