Integrative Immunobiology Section
Established in 2008
Stefan Muljo, Ph.D.
Chief, Integrative Immunobiology Section

Major Areas of Research
- RNA-binding proteins and regulatory non-coding RNAs: characterization under physiological and pathological conditions, regulation of production, mechanisms of action, identification of cognate targets
- Gene expression programs and their regulation in hematopoietic stem cells and during cellular differentiation
- Targeting microRNAs for modulating or enhancing immune responses
- Post-transcriptional regulation of human endogenous retroviruses (HERVs)
- In utero hematopoietic stem cell transplantation
- In vivo function of innate-like B-1 cells
Program Description
The Integrative Immunobiology Section is interested in understanding how gene expression programs are orchestrated as cells differentiate from stem cells into various lineages in the immune system. In particular, our team of investigators studies how these systems are controlled by microRNAs, long non-coding RNAs, and RNA-binding proteins. Our major goal is to discover novel molecular circuits that control cell fates in the hematopoietic and immune systems, since perturbations in their genetic programming underlie many diseases including cancer, immunodeficiency, autoimmunity, inflammation, allergy and infectious diseases. To address these fundamental issues, we employ an integrative systems biology approach to reverse engineer the molecular logic of cellular differentiation. We combine genome- and transcriptome-wide measurements with experimental perturbations in order to test and refine our models. As one example of our success, we identified the RNA-binding protein Lin28b as a lineage-determining factor for fetal hematopoietic stem cells, a discovery with potentially important implications for Regenerative Medicine. At a basic level, we demonstrate the concept of post-transcriptional (re)programming of cell fate decisions.
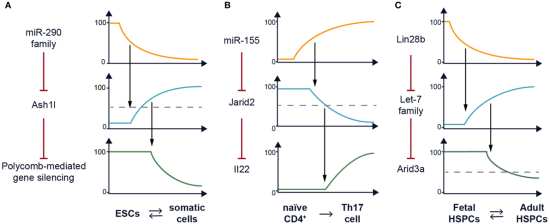
Recurrent network motif in post-transcriptional (re)programming. This common theme emerged from the work of our section over its first 10 years at NIAID. These three examples from our short history demonstrate how post-transcriptional regulation can alter gene expression programs dramatically. Adapted from Posttranscriptional (Re)programming of Cell Fate: Examples in Stem Cells, Progenitor, and Differentiated Cells.
What is Integrative Immunobiology? Our modus operandi is to integrate data obtained from orthogonal but complementary approaches (depicted in the following figure).
Synopsis of integrative immunobiology
Primarily, we wish to understand how the immune system develops and functions. We study this from the perspective of RNA biology because it is interesting and provides many opportunities. We take advantage of cutting-edge technologies such as genome editing and next generation sequencing. In addition, we couple our studies with traditional and modern molecular and cell biology techniques which are constantly improving (e.g, spatial transcriptomics). Finally, we use high performance computing to analyze, organize and integrate all these data to generate a working model of the system that we are studying. Ultimately, we want to provide novel insights that can be translated to improve human health. We have a long-standing interest in hematopoietic stem cells, the “poster child” of Regenerative Medicine. We have ideas for novel applications of gene and cell therapies particularly to correct inborn errors of immunity and hematopoiesis (e.g., sickle cell disease and beta-thalassemia). We have a pending patent application with Dr. Jeffrey Miller (NIDDK) for a Method of Increasing the Amount of Fetal Hemoglobin in a Cell and/or Mammal (# PCT/US2013/067811) that could be developed to treat beta-globinopathies.
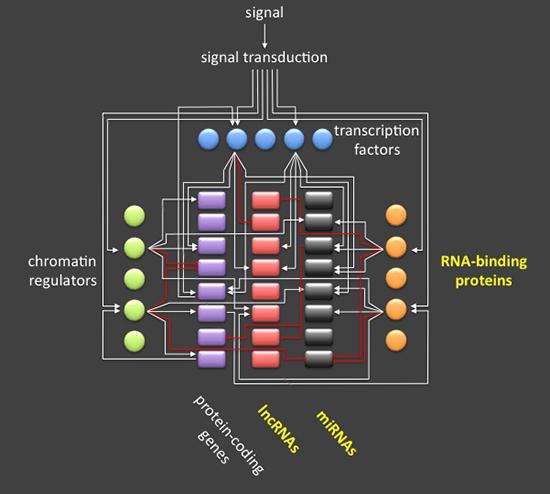
Models of gene regulatory networks need to integrate RNA-binding proteins, long non-coding RNAs (lncRNAs), and microRNAs (miRNAs) in order to be more accurate and predictive. While it is well established that gene expression programs and cell fates are orchestrated by signaling, epigenetic and transcription factors, we believe that post-transcriptional (re)programming is also an important area of research.
A cell's fate is largely determined by its transcriptome and proteome which are regulated by transcriptional and post-transcriptional networks. Here they are depicted as an integrated circuit that processes input (signal) to mediate an output (not depicted). For simplicity, post-translational, metabolic, and other molecular networks are not depicted here. Chromatin regulators and transcription factors with the aid of lncRNAs control the accessibility and transcription rate of protein-coding and non-coding genes while RNA-binding proteins collaborate with non-coding RNAs to orchestrate the processing, transport, translation, and stability of RNA transcripts. Post-transcriptional regulation has evolved an important additional layer that shapes the transcriptome in response to developmental and environmental cues that may not be achievable by transcriptional regulation alone. Adapted from Exploring the RNA world in hematopoietic cells through the lens of RNA-binding proteins.
Many laboratories are studying protein-coding genes. Although proteins are clearly important, protein-coding genes only constitute less than five percent of the human genome. Thus, what might the remaining 95 percent be doing? Previously, it was called “junk DNA.” Intriguingly, approximately 75 percent of the human genome is transcribed and potentially there are transcripts that may offer yet unappreciated functions. For instance, microRNAs were not discovered until 1993. In order to systematically study the “non-coding” genome and transcriptome, we will need to invest in functional genomics. While the sequencing of genomes has provided us with comprehensive parts lists (i.e., genotypes), we need to figure out the function of all these parts. Furthermore, we need systems biology to begin to understand how different parts are coming together to generate emergent properties (i.e., phenotypes).
Biography
Education
Ph.D., The Johns Hopkins University School of Medicine, Baltimore, MD
Dr. Muljo earned his Ph.D. from the Graduate Program in Immunology at The Johns Hopkins University School of Medicine. Part of his dissertation work was performed at the department of molecular and cell biology in the division of immunology and pathogenesis, University of California, Berkeley. This was followed by a postdoctoral fellowship at the Immune Disease Institute (formerly the Center for Blood Research), Harvard Medical School. He was recruited to the Laboratory of Immunology (LI) in 2008 as a tenure-track investigator. In 2016, he was promoted to tenured Senior Investigator and is head of the Integrative Immunobiology Section, which is now part of the Laboratory of Immune System Biology (LISB). He is a faculty member of the NIH-Penn, and NIH-OxCam graduate partnership programs, as well as others.
Special Interest Groups
- Immunology
- Systems biology
- Genetics
- Stem cell
- RNA
- Transcription factors
- Cytokine
- Single cell genomics
Selected Publications
Foroushani AK, Chim B, Wong M, Rastegar A, Smith PT, Wang S, Barbian K, Martens C, Hafner M, Muljo SA. Posttranscriptional regulation of human endogenous retroviruses by RNA-binding motif protein 4, RBM4. Proc Natl Acad Sci U S A. 2020 Oct 20;117(42):26520-26530.
Wang S, Chim B, Su Y, Khil P, Wong M, Wang X, Foroushani A, Smith PT, Liu X, Li R, Ganesan S, Kanellopoulou C, Hafner M, Muljo SA. Enhancement of LIN28B-induced hematopoietic reprogramming by IGF2BP3. Genes and Development. 2019; 33: 1048–10688.
Kanellopoulou C, Gilpatrick T, Kilaru G, Burr P, Nguyen CK, Morawski A, Lenardo MJ, Muljo SA. Reprogramming of Polycomb-mediated gene silencing in embryonic stem cells by the miR-290 family and the methyltransferase Ash1l. Stem Cell Reports. 2015; 5: 971–978.
Escobar TM, Kanellopoulou C, Kugler DG, Kilaru G, Nguyen CK, Nagarajan V, Bhairavabhotla R, Northrup D, Zahr R, Burr P, Liu X, Zhao K, Sher A, Jankovic D, Zhu J, Muljo SA. miR-155 activates cytokine gene expression in Th17 cells by regulating the DNA-binding protein Jarid2 to relieve Polycomb-mediated repression. Immunity. 2014; 40: 865-879.
Yuan J, Nguyen CK, Liu X, Kanellopoulou C, Muljo SA. Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis. Science. 2012; 335:1195-1200.
Useful Datasets
| NCBI GEO | Description | Samples | Species |
|---|---|---|---|
| GSE35214 | NanoString microRNA expression profiles | 4 | Mus musculus |
| GSE34854 | RNA-seq of thymocytes ± Lin28 | 2 | Mus musculus |
| GSE47528 | ChIP-seq of Th17 cells | 12 | Mus musculus |
| GSE37228 | RNA-seq of WT vs miR-155 KO Th17 cells | 2 | Mus musculus |
| GSE69304 | ATF4 ChIP-seq in HAP1 cell line | 4 | Homo sapiens |
| GSE69308 | RNA-seq of WT HAP1 cells vs ATF4 KO | 4 | Homo sapiens |
| GSE58766 | RNA-seq of DT40 cell line | 1 | Gallus gallus |
| GSE60397 | ChIP-seq and RNA-seq of WT vs Dicer1 KO ES cells | 16 | Mus musculus |
| GSE148267 | RNA-seq of Treg vs Tconv | 12 | Homo sapiens |
| GSE98429 | RNA-seq of 220-8 pro-B cells ± LIN28 | 2 | Mus musculus |
| GSE98430 | RNA-seq of fetal liver vs bone marrow HSPCs | 4 | Mus musculus |
| GSE98465 | LIN28, Igf2bp3 and Pum2 PAR-CLIP in 220-8 cells | 13 | Mus musculus |
| GSE108129 | Single-cell RNA-seq of bone marrow CLPs | 4 | Mus musculus |
| GSE138063 | IGF2BP1 and IGF2BP3 PAR-CLIP in K562 cell line | 4 | Homo sapiens |
| GSE128308 | RNA-seq of Th17 cells + FGIN-1-27 or DMSO | 6 | Mus musculus |
| GSE147893 | RNA-seq of WT HAP1 cells vs RBM4 KO | 10 | Homo sapiens |
| GSE147895 | FLAG-RBM4 PAR-CLIP in HAP1 cells | 8 | Homo sapiens |
| GSE147896 | PacBio RNA-seq of WT HAP1 cells vs RBM4 KO | 2 | Homo sapiens |
Research Networks
- NIH Oxford-Cambridge Scholars Program
- NIH-Penn Immunology Graduate Partnership Program
- NIH-JHU Graduate Partnership Program
Fellowship
We are dedicated to training the next generation of biomedical researchers. A joint doctoral studentship position is available in partnership with Dr. Anindita Roy’s laboratory to study fetal hematopoiesis in mice at NIH, and in humans at Oxford University, United Kingdom, as part of the NIH-OxCam Partnership Program. For instructions on how to apply for the program, see Graduate Partnerships Program.
Haematopoiesis is a finely tuned process by which mature blood cells of multiple lineages are constantly generated throughout life from haematopoietic stem cells. In humans, definitive haematopoiesis commences in the fetal liver at around five weeks of gestation, and remains the main site of haematopoiesis throughout fetal life. Haematopoiesis in the bone marrow starts around 11-12 weeks of gestation, but does not take over as the primary site of haematopoiesis until just after birth. Recent evidence suggests that fetal haematopoiesis is distinct from postnatal haematopoiesis in many ways. Most of these studies have been done on mouse models, but whether these differences exist in, or are a true reflection of haematopoiesis in the human setting, remains to be determined. We, and others have begun to investigate unique features of human fetal haematopoiesis and this project will determine fetal specific programmes that change through ontogeny. Studying haematopoiesis throughout the human lifespan may be important not only to understand normal developmental processes, but also to understand the pathogenesis of postnatal haematological diseases that may have their origins in fetal life. It may also have therapeutic implications in the use of haematopoietic stem cells from donors of different ages as these cells vary in their composition and differentiation potential, which may depend on the physiological processes or demands of that particular developmental stage, and/or in response to specific microenvironmental cues.
Videos
Roles of regulatory RNAs & RNA-binding proteins in (re)programming patterns of gene expression
Research Group
Our interdisciplinary team studies how the immune system develops and functions through the lens of RNA biology. We are interested in learning how gene expression programs are regulated post-transcriptionally with the goal of manipulating cell fate.